Construction of a simple pipeline to run BLAST jobs¶
Overview¶
This is a simple example to illustrate the convenience Ruffus brings to simple tasks in bioinformatics.
- Split a problem into multiple fragments that can be
- Run in parallel giving partial solutions that can be
- Recombined into the complete solution.
The example code runs a ncbi blast search for four sequences against the human refseq protein sequence database.
- Split each of the four sequences into a separate file.
- Run in parallel Blastall on each sequence file
- Recombine the BLAST results by simple concatenation.
In real life,
- BLAST already provides support for multiprocessing
- Sequence files would be split in much larger chunks, with many sequences
- The jobs would be submitted to large computational farms (in our case, using the SunGrid Engine).
- The High Scoring Pairs (HSPs) would be parsed / filtered / stored in your own formats.
Note
This bioinformatics example is intended to showcase some of the features of Ruffus.
- See the manual to learn about the various features in Ruffus.
Prerequisites¶
1. Ruffus¶
To install Ruffus on most systems with python installed:
easy_install -U ruffusOtherwise, download Ruffus and run:
tar -xvzf ruffus-xxx.tar.gz cd ruffus-xxx ./setup installwhere xxx is the latest Ruffus version.
2. BLAST¶
3. human refseq sequence database¶
We also need to download the human refseq sequence file and format the ncbi database:
wget ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/mRNA_Prot/human.protein.faa.gz gunzip human.protein.faa.gz formatdb -i human.protein.faa
4. test sequences¶
Query sequences in FASTA format can be found in original.fa
Step 1. Splitting up the query sequences¶
We want each of our sequences in the query file original.fa to be placed in a separate files named
XXX.segmentwhereXXX= 1 -> the number of sequences.current_file_index = 0 for line in open("original.fa"): # start a new file for each accession line if line[0] == '>': current_file_index += 1 current_file = open("%d.segment" % current_file_index, "w") current_file.write(line)To use this in a pipeline, we only need to wrap this in a function, “decorated” with the Ruffus keyword @split:
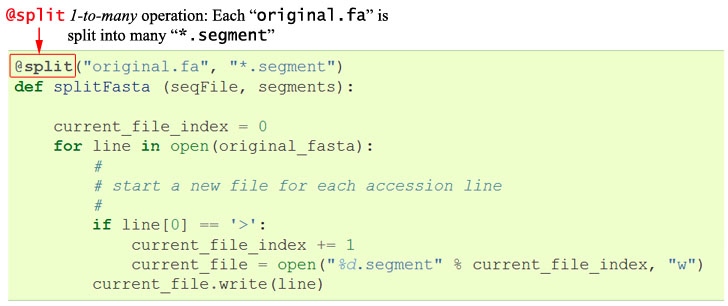 This indicates that we are splitting up the input file original.fa into however many
This indicates that we are splitting up the input file original.fa into however many*.segmentfiles as it takes.The pipelined function itself takes two arguments, for the input and output.We shall see later this simple @split decorator already gives all the benefits of:
- Dependency checking
- Flowchart printing
Step 2. Run BLAST jobs in parallel¶
Assuming that blast is already installed, sequence matches can be found with this python code:
os.system("blastall -p blastp -d human.protein.faa -i 1.segment > 1.blastResult")To pipeline this, we need to simply wrap in a function, decorated with the Ruffus keyword @transform.
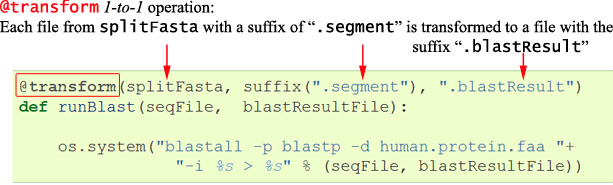
This indicates that we are taking all the output files from the previous
splitFastaoperation (*.segment) and @transform-ing each to a new file with the.blastResultsuffix. Each of these transformation operations can run in parallel if specified.
Step 3. Combining BLAST results¶
- The following python code will concatenate the results together
To pipeline this, we need again to decorate with the Ruffus keyword @merge.
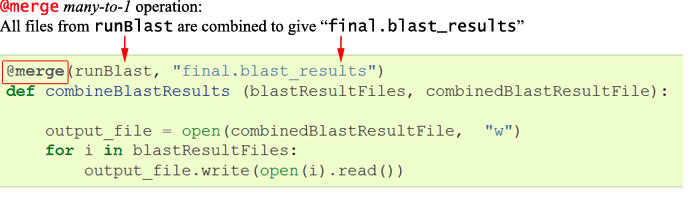
This indicates that we are taking all the output files from the previous
runBlastoperation (*.blastResults) and @merge-ing them to the new filefinal.blast_results.
Step 4. Running the pipeline¶
We can run the completed pipeline using a maximum of 4 parallel processes by calling pipeline_run :
pipeline_run([combineBlastResults], verbose = 2, multiprocess = 4)Though we have only asked Ruffus to run
combineBlastResults, it traces all the dependencies of this task and runs all the necessary parts of the pipeline.Note
The full code for this example can be found here suitable for pasting into the python command shell.
The
verboseparameter causes the following output to be printed to stderr as the pipeline runs:>>> pipeline_run([combineBlastResults], verbose = 2, multiprocess = 4) Job = [original.fa -> *.segment] completed Completed Task = splitFasta Job = [1.segment -> 1.blastResult] completed Job = [3.segment -> 3.blastResult] completed Job = [2.segment -> 2.blastResult] completed Job = [4.segment -> 4.blastResult] completed Completed Task = runBlast Job = [[1.blastResult, 2.blastResult, 3.blastResult, 4.blastResult] -> final.blast_results] completed Completed Task = combineBlastResults
Step 5. Testing dependencies¶
If we invoked pipeline_run again, nothing further would happen because the pipeline is now up-to-date. But what if the pipeline had not run to completion?
We can simulate the failure of one of the
blastalljobs by deleting its results:os.unlink("4.blastResult")Let us use the pipeline_printout function to print out the dependencies of the pipeline at a high
verboselevel which will show both complete and incomplete jobs:>>> import sys >>> pipeline_printout(sys.stdout, [combineBlastResults], verbose = 4) ________________________________________ Tasks which are up-to-date: Task = splitFasta "Split sequence file into as many fragments as appropriate depending on the size of original_fasta" ________________________________________ Tasks which will be run: Task = runBlast "Run blast" Job = [4.segment ->4.blastResult] Job needs update: Missing file 4.blastResult Task = combineBlastResults "Combine blast results" Job = [[1.blastResult, 2.blastResult, 3.blastResult, 4.blastResult] ->final.blast_results] Job needs update: Missing file 4.blastResult ________________________________________Only the parts of the pipeline which involve the missing BLAST result will be rerun. We can confirm this by invoking the pipeline.
>>> pipeline_run([combineBlastResults], verbose = 2, multiprocess = 4) Job = [1.segment -> 1.blastResult] unnecessary: already up to date Job = [2.segment -> 2.blastResult] unnecessary: already up to date Job = [3.segment -> 3.blastResult] unnecessary: already up to date Job = [4.segment -> 4.blastResult] completed Completed Task = runBlast Job = [[1.blastResult, 2.blastResult, 3.blastResult, 4.blastResult] -> final.blast_results] completed Completed Task = combineBlastResults
What is next?¶
In the next (short) part, we shall add some standard (boilerplate) code to turn this BLAST pipeline into a (slightly more) useful python program.