Part 2: A slightly more practical pipeline to run blasts jobs¶
Overview¶
Previously, we had built a simple pipeline to split up a FASTA file of query sequences so that these can be matched against a sequence database in parallel.
We shall wrap this code so that
- It is more robust to interruptions
- We can specify the file names on the command line
Step 1. Cleaning up any leftover junk from previous pipeline runs¶
We split up each of our sequences in the query file original.fa into a separate files namedXXX.segmentwhereXXXis the number of sequences in the FASTA file.However, if we start with 6 sequences (giving1.segment...6.segment), and we then edited original.fa so that only 5 were left, the file6.segmentwould still be left hanging around as an unwanted, extraneous and confusing orphan.As a general rule, it is a good idea to clean up the results of a previous run in a @split operation:
@split("original.fa", "*.segment") def splitFasta (seqFile, segments): # # Clean up any segment files from previous runs before creating new one # for i in glob.glob("*.segment"): os.unlink(i) # code as before...
Step 2. Adding a “flag” file to mark successful completion¶
When pipelined tasks are interrupted half way through an operation, the output may only contain part of the results in an incomplete or inconsistent state. There are three general options to deal with this:
- Catch any interrupting conditions and delete the incomplete output
- Tag successfully completed output with a special marker at the end of the file
- Create an empty “flag” file whose only point is to signal success
Option (3) is the most reliable way and involves the least amount of work in Ruffus. We add flag files with the suffix
.blastSuccessfor our parallel BLAST jobs:@transform(splitFasta, suffix(".segment"), [".blastResult", ".blastSuccess"]) def runBlast(seqFile, output_files): blastResultFile, flag_file = output_files # # Existing code unchanged # os.system("blastall -p blastp -d human.protein.faa "+ "-i %s > %s" % (seqFile, blastResultFile)) # # "touch" flag file to indicate success # open(flag_file, "w")
Step 3. Allowing the script to be invoked on the command line¶
We allow the query sequence file, as well as the sequence database and end results to be specified at runtime using the standard python optparse module. We find this approach to run time arguments generally useful for many Ruffus scripts. The full code can be viewed here and downloaded from run_parallel_blast.py.
The different options can be inspected by running the script with the
--helpor-hargument.The following options are useful for developing Ruffus scripts:
--verbose | -v : Print more detailed messages for each additional verbose level. E.g. run_parallel_blast --verbose --verbose --verbose ... (or -vvv) --jobs | -j : Specifies the number of jobs (operations) to run in parallel. --flowchart FILE : Print flowchart of the pipeline to FILE. Flowchart format depends on extension. Alternatives include (".dot", ".jpg", "*.svg", "*.png" etc). Formats other than ".dot" require the dot program to be installed (http://www.graphviz.org/). --just_print | -n Only print a trace (description) of the pipeline. The level of detail is set by --verbose.
Step 4. Printing out a flowchart for the pipeline¶
The
--flowchartargument results in a call topipeline_printout_graph(...)This prints out a flowchart of the pipeline. Valid formats include ”.dot”, ”.jpg”, ”.svg”, ”.png” but all except for the first require thedotprogram to be installed (http://www.graphviz.org/).The state of the pipeline is reflected in the flowchart:
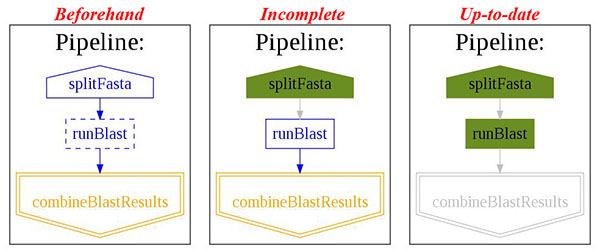
Step 5. Errors¶
Because Ruffus scripts are just normal python functions, you can debug them using your usual tools, or jump to the offending line(s) even when the pipeline is running in parallel.
For example, these are the what the error messages would look like if we had mis-spelt
blastal. In run_parallel_blast.py, python exceptions are raised if theblastallcommand fails.Each of the exceptions for the parallel operations are printed out with the offending lines (line 204), and problems (
blastalnot found) highlighted in red.
Step 6. Will it run?¶
The full code can be viewed here and downloaded from run_parallel_blast.py.